Vedoucí výzkumného programu: Vladimír Kryštof

Roskovitin a jeho pyrazolopyrimidinový isoster v aktivním místě CDK2. Převzato z J. Med. Chem. 2011, 54, 2980–2993.
Fosforylace protein představuje univerzální mechanismus regulace aktivity protein zapojených prakticky ve všech buněčných procesech, včetně proliferace a buněčného cyklu. Cyklin-dependentní kinasy (CDK) jsou enzymy, které hrají klíčovou roli v rozhodování o vstupu do buněčného cyklu (CDK4 a CDK6), ovládají replikaci DNA (CDK2) a zahajují mitózu (CDK1). Některé další enzymy z rodiny CDK se podílejí na odlišných procesech, jako je transkripce mRNA a její sestřih, apoptóza, buněčná motilita, sebeobnovování kmenových buněk, spermatogeneze, funkčnost neuronů.
V nádorech byly identifikovány četné abnormality vztahující se k aktivitám některých CDK. Mezi nejčastější patří mutace nebo umlčování exprese inhibitoru p16INK4A, mutace RB1 nebo jeho inaktivace na proteinové úrovni, deregulace cyklinů třídy D, amplifikace genu kódujícího CDK4 nebo cykliny třídy E. Identifikace těchto molekulárních změn iniciovalo vývoj nízkomolekulárních inhibitorů CDK, které proliferaci blokují a indukují smrt nádorových buněk. Jejich využití v buněčné biologii a experimentální onkologii se přímo nabízelo.
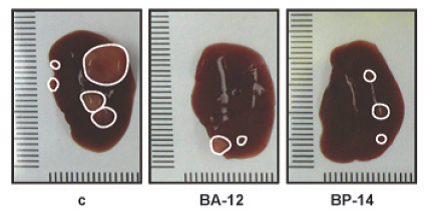
Reprezentativní morfologie endogenního karcinomu jater myši neléčené (c) a léčené dvěma inhibitory CDK. Převzato z Mol Cancer Ther 2013;12:1947-1957.
Spolu s postupným nárůstem poznatků o funkcích CDK jak v normálních tak v transformovaných buňkách byly po celém světě zahajovány výzkumné program, směřující k vývoji inhibitorů jako protinádorových léčiv. Také v naší laboratoři byly připraveny a charakterizovány některé inhibitory CDK, a to zejména na bázi purinu a jemu isosterních heterocyklických systémů, jako jsou 8-azapuriny, pyrazolo[4,3-d]pyrimidiny nebo pyrazolo[4,3-e][1,2,4]triaziny. Nejúčinnější sloučeniny vykazují vynikající účinky nejen v biochemických experimentech, ale take na buněčné úrovni či v myších modelech. Jejich protinádorová účinnost přitom není dána jen negativním vlivem na proliferaci nádorových buněk. Nedávno jsme totiž zjistili, že inhibice CDK5 může mít rovněž významný protinádorový vliv, a to v kontextu nádorové angiogeneze.
Naše sloučeniny jsou schopny zastavovat proliferaci cévních endotelových buněk, blokovat jejich migraci a dokonce výrazně omezovat tvorbu nových cév ve zvířecích modelech. Tyto projevy jsou nejspíše dány právě inhibicí CDK5 v endotelových buňkách.
Nedávno jsme vyvinuli take nové inhibitory receptorové kinasy FLT3. Kinasa FLT3 patří mezi významné cíle léčiv pro akutní myeloidní leukemii (AML), neboť je u řady pacientů aktivovaná mutací ITD. Naše inhibitory vykazují nanomolární aktivitu v biochemických a buněčných experimentech a vysoce selektivně zastavují proliferaci buněk AML s mutací FLT3-ITD. V leukemických buňkách blokují aktivitu FLT3 a podřízené signální dráhy. Velmi významný protinádorový účinek byl prokázán také v myším modelu AML.
Nízkomolekulární inhibitory proteinkinas proto považujeme za velmi atraktivní skupinu potenciálních protinádorových léčiv s vícenásobným mechanismem účinku. Vývoj a charakterizace inhibitorů proteinkinas na méně probádaných heterocyklických systémech je proto jedním z našich současných výzkumných záměrů.
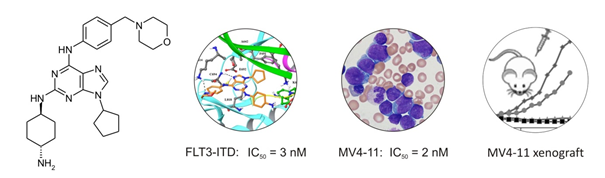 |
| Kandidátní inhibitor onkogenní kinasy FLT3 vykazuje nanomolární účinnost v biochemických a buněčných experimentech a také terapeutický vliv v myším modelu. Převzato z J Med Chem. 2018 May 10;61(9):3855-3869. |